

Our research
The research team deals with quantum-chemical investigation of selected properties of mainly organic molecules as well as with the development of methods for their calculation.
First, we are interested in spin-dependent relativistic effects like spin-orbit coupling, spin-spin coupling or parity-violation coupling. These are responsible for various phenomena comprising zero-field splitting, radiative and radiationless spin-forbidden transitions or tiny differences between electronic-state energies of enantiomers. The focus is on predicting photophysical and photochemical properties of synthetically or biologically relevant molecules, unraveling reaction mechanisms involving more than one potential energy surface, and on designing molecules having appealing, often anomalous, properties (e.g. those showing inverse heavy-atom effect on the rate of spin-forbidden transitions or those with an extraordinarily large ground-state energy shift due to parity violation). Most of the calculations are carried out on biradicals (reactive intermediates with nearly degenerate lowest singlet and triplet states) and transition-metal complexes.
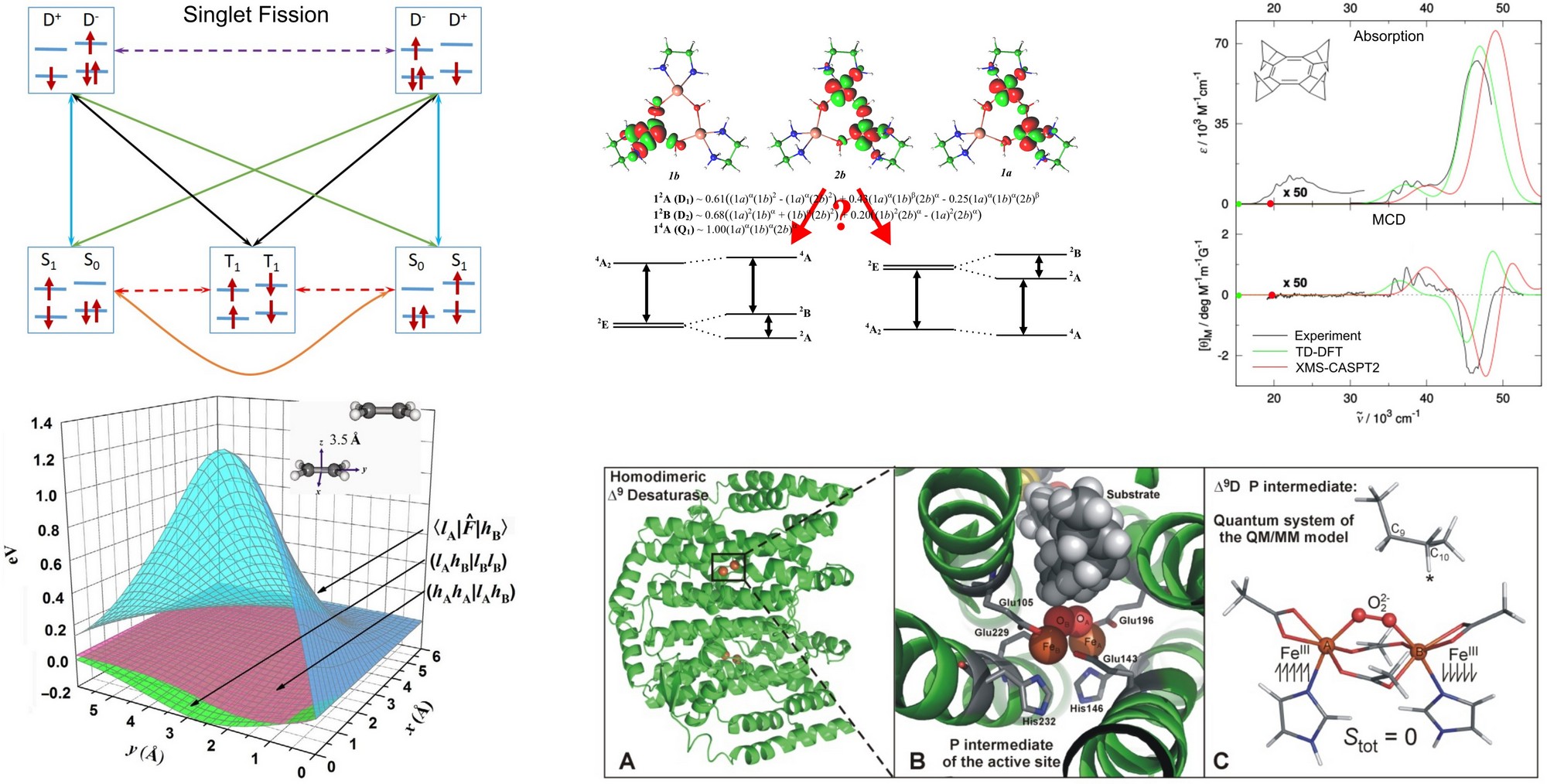
Next, the team is involved in the project concerning the improvement of the efficiency of the titanium dioxide based solar cells. We study the possibility of singlet fission mechanism of excited organic dyes. After conversion of the first excited singlet state of a dye into a pair of triplet, the dye can, in principle, inject two electrons into the crystalline titanium dioxide thus enhancing the efficiency of the solar cell by a factor of two. We use simple as well as sophisticated quantum chemical calculations of exited singlet and triplet states to design the structure of a dye, which is afterwards synthesized and measured in collaboration with the experimental groups in the Institute.
Last but not least, we are also engaged in predicting the pKa values of nucleotides in their aqueous solutions. We use the method COSMO-RS, the combination of the quantum chemical dielectric continuum solvation model COSMO with a statistical thermodynamics treatment for realistic solvation simulations.